Microglia Give Astrocytes License to Kill
Quick Links
In their healthy state, astrocytes busy themselves nursing neurons—forming and pruning synapses, clearing used neurotransmitters, and delivering nutrients. Now, a paper in the January 18 Nature reports that three cytokines released by microglia turn these helpers into killers. Scientists led by Ben Barres and Shane Liddelow, Stanford University School of Medicine, Palo Alto, California, find that Il-1α, TNF, and C1q push resting astrocytes into a reactive state the scientists call A1. A1 astrocytes weaken synapses and kill neurons and myelin-producing cells. The authors find A1 astrocytes in a variety of injuries and disease states, including Alzheimer’s, and suggest they are at the root of neuron death in these cases. By simply blocking secretion or action of those three microglial cytokines to keep astrocytes out of the A1 state, it may be possible to treat these disorders, they suggest. “Acute injuries of the retina, brain, and spinal cord, and even neurodegenerative diseases, may be more treatable than we thought,” Liddelow said.
“This article is really exciting,” wrote Jonathan Kipnis, University of Virginia School of Medicine, Charlottesville, to Alzforum. “It’s one of those works where you say, how come we did not think about this before?”
Philip Haydon, Tufts University, Boston, agreed, saying that in the past, scientists thought that astrocytes became reactive in response to neuronal pathology. “Now they are showing that astrocytes change first, and that dramatically affects synapses and neurodegeneration. This almost turns everything upside down.”
“We’ve known about reactive astrocytes in disease for a long time, but why and when astrocytes become reactive and how they behave has been a mystery,” said Cagla Eroglu, Duke University, Durham, North Carolina. “This paper marks a milestone toward understanding that. If we study, understand, and target this specific population of cells, it might be possible to help with not just one, but many diseases of the nervous system.”
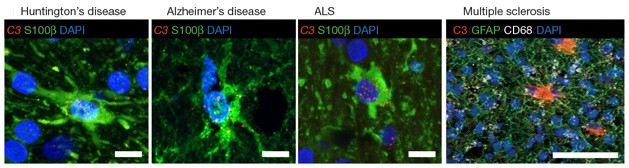
Common Theme: In various diseases, astrocytes (green) are converted to the A1 state, in which they release complement factors such as C3 (red). [Liddelow et al., 2016. Nature.]
In a previous study, Barres’ group reported two forms of reactive astrocytes, characterized by different transcriptome profiles (Zamanian et al., 2012). Systemically injecting the bacterial lipopolysaccharide (LPS) into mice induced the A1 state, where astrocytes began to spew synapse-harming classical complement cascade genes, such as C3. Recent research by Beth Stevens, Children’s Hospital, Boston, a co-author on the current paper, suggests complement proteins play a role in synaptic pruning in Alzheimer’s disease (Apr 2016 news). In ischemic stroke, on the other hand, astrocytes assume a helpful A2 state, and release neurotrophins. The researchers wondered how LPS could bring about the A1 state, as astrocytes have no receptors for the molecule. Could microglia mediate the response? After all, these cells do have the necessary receptors and are activated by LPS.
To find out, Liddelow and colleagues injected mice with LPS, then measured gene expression to determine the state of astrocytes. A1 astrocytes quickly sprung up in control mice, but none developed if animals were genetically engineered to lack microglia. That suggested the response hinged on microglia. But how? A screen of the signals secreted by microglia turned up Il-1α, TNF, and C1q, each of which coaxed astrocytes partway toward an A1 phenotype. Together, the three made A1 cells nearly identical to those induced by LPS in vivo. Previous studies have hinted that microglia-secreted factors induce reactive astrocytes (Kyrkanides et al., 1999).
Could taking them away prevent A1 formation? Yes, it seems. Genetically knocking out each cytokine led to fewer LPS-induced A1 astrocytes in culture; ablating all three prevented them completely. Once created, however, removing the cytokines with antibodies did not appear to reverse the A1 phenotype, which persisted for at least a week. Treating the cells with anti-inflammatory cytokines TGFβ or FGF did suppress A1 transcript levels to some degree. “That gives us great hope moving forward that this could be druggable,” said Liddelow.
Once astrocytes morph into A1s, how does their function change? The scientists cultured mouse retinal ganglion cells (RGCs) with either resting or A1 astrocytes and compared the number of new synapses by staining for pre- and postsynaptic proteins. Only half as many synapses formed in the presence of A1 astrocytes, which also suppressed RGC firing and weakened their action potentials. While resting astrocytes prune synapses and clear debris, A1 astrocytes engulfed 50 to 75 percent fewer synaptosomes and phagocytosed almost no myelin. In mice, too, A1 astrocytes consumed only half the normal amount of synaptosomes.
Co-culture with A1 astrocytes proved fatal for RGCs, cortical neurons, embryonic spinal motor neurons, and myelin-producing oligodendrocytes. A1 cells killed a quarter of dopaminergic neurons and slowed the differentiation of oligodendrocyte precursors. Curiously, certain motor neurons escaped unscathed. Together, the results suggested that A1 astrocytes secrete a mystery factor toxic to certain CNS neurons and mature oligodendrocytes. Liddelow and colleagues are now trying to find this A1-secreted death signal.
They are also working to induce the helpful A2 state. A drug that prevented A1 formation could prevent neurodegeneration, stimulate synapse growth, and clear debris, the authors wrote. FDA-approved antibodies against Il-1α and TNF used for other diseases could be tested quickly in AD, said Liddelow.
Role in Disease
Reactive astrocytes turn up in numerous diseases and injuries in which neurons die, though their form and function remains unclear. The reason for neuronal death in these situations is unknown. In the case of physical trauma, the authors blame A1 astrocytes. When Liddelow crushed the axons of retinal neurons in rats or mice, A1 astrocytes surrounded the damage and two-thirds of the neurons died after a week. However, when he also injected anti-Il-1α, -TNF, and -C1q antibodies, or genetically deleted the genes for those three cytokines, no A1 astrocytes formed and RCGs survived the entire one- and two-week experiments. These findings hint that an anti-A1 treatment could prevent neuron death in the case of injury.
Do activated microglia spawn A1 astrocytes in neurodegenerative disease? The authors looked for C3, a marker of A1 activation, in astrocytes from human tissue affected by Alzheimer’s, Huntington’s, Parkinson’s, amyotrophic lateral sclerosis, and multiple sclerosis. In each, the most affected brain regions were filled with A1 astrocytes (see image above). For instance, nearly 60 percent of the astrocytes in the prefrontal cortices of AD patients’ brains tested positive for C3. Liddelow speculated that this astrocyte reactivity was driving neurodegeneration.
“This combination of in vitro and in vivo experiments, then validation in human tissue, makes this such a powerful study,” Haydon said. “A key factor missing in our understanding of neurodegeneration is why nerve cells are dying,” said Brian MacVicar, University of British Columbia, Vancouver, Canada. “It is crystal clear here that it involves an initial inflammatory response from microglial cells that then triggers a toxic phenotype in astrocytes.”
Kerry O’Banion, University of Rochester Medical Center, New York, noted that the authors found A1 astrocytes in human tissue, but stopped short of showing if and how they were toxic. The data hint at the intriguing possibility that they are. “If you can inhibit the toxicity, or reduce the prevalence, of A1 astrocytes, then you might be able to slow disease progression,” he told Alzforum. He cautioned that to be an effective treatment target, this process would need to be important early on in disease. If it only happens at advanced stages, preventing it may not help, he said.
“This is an important and thought-provoking study,” said Michael Sofroniew, University of California, Los Angeles. “It will give us specific molecular targets we can try to manipulate to reduce neurodegeneration.” However, Sofroniew cautioned that a signaling response this specific and conserved across species likely has a purpose. He wondered whether this mechanism is getting rid of already defective neurons. “If this process is only activated when it’s time to call in the undertaker, not the ambulance, then stopping it may not be helpful. You might be left with something non-functional.”—Gwyneth Dickey Zakaib
References
News Citations
Paper Citations
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012 May 2;32(18):6391-410. PubMed.
- Kyrkanides S, Olschowka JA, Williams JP, Hansen JT, O'Banion MK. TNF alpha and IL-1beta mediate intercellular adhesion molecule-1 induction via microglia-astrocyte interaction in CNS radiation injury. J Neuroimmunol. 1999 Mar 1;95(1-2):95-106. PubMed.
Further Reading
Papers
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010 Jan;119(1):7-35. Epub 2009 Dec 10 PubMed.
Primary Papers
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017 Jan 26;541(7638):481-487. Epub 2017 Jan 18 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.